Neuromyelitis Optica
NMOSD, distinct from MS, is marked by AQP4 antibodies causing optic neuritis and myelitis. Diagnosis involves specific clinical criteria and antibody presence. Treatment includes methylprednisolone, plasma exchange, and immunotherapies like eculizumab and rituximab. Refractory cases may benefit from azathioprine or mycophenolate mofetil, with therapy typically lasting five years.
Neuromyelitis Optica Spectrum Disorders (NMOSD) Overview:
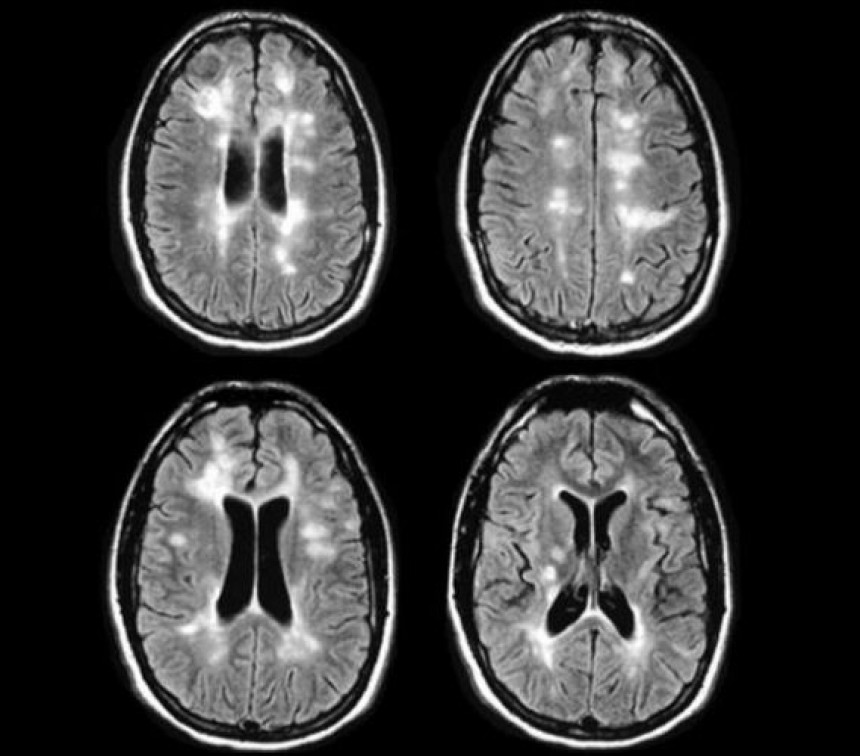
Neuromyelitis optica spectrum disorders (NMOSD), previously known as neuromyelitis optica [NMO] are inflammatory disorders of the central nervous system characterised by immune- mediated severe demyelination which causes axonal damage in optic nerves and the spinal cord. (1)
NMOSD Distinction from MS:
NMOSD is now recognised as a distinct clinical entity from MS with the discovery of a disease specific serum NMO- immunoglobulin G (IgG) antibody. NMO IgG selectively binds aquaporin- 4(AQP4) which is highly concentrated in the spinal cord, periaqueductal and periventricular regions of the CNS. Unlike MS, necrosis and cavitation typically involve both grey and white matter. Also, NMOSD is characterised by humoral immune response rather than cell mediated response as in MS. (2)
Clinical Presentation of NMOSD:
NMOSD usually presents as acute attacks of bilateral or rapidly sequential optic neuritis, leading to severe visual loss or as transverse myelitis, causing limb weakness, sensory loss, and bladder dysfunction with typically relapsing courses. Also, some patients with NMOSD present with brainstem symptoms due to medullary involvement. Brainstem involvement may lead to acute respiratory failure and death. (3, 4)
Diagnostic Criteria for NMOSD:
Diagnostic criteria (Revised consensus criteria for the diagnosis of NMOSD) is based on the presence of at least one core clinical characteristics, a positive AQP4 antibody status and exclusion of an alternative diagnosis (MRI features). There are six core clinical characteristics, which are optic neuritis, acute transverse myelitis, area postrema syndrome (episode of unexplained hiccups, nausea and vomiting), acute brainstem syndrome, symptomatic narcolepsy and symptomatic cerebral syndrome with NMOSD typical brain lesions. (5)
Seronegative NMOSD and MOG Antibodies:
If AQP4-IgG antibody is negative, then for diagnosis of NMOSD, there should be at least 2 core clinical characteristics, with at least one core clinical characteristic must be optic neuritis, acute transverse myelitis with longitudinally extensive or area postrema syndrome with dissemination in space and fulfilment of additional MRI requirements. (6)
Treatment of Acute NMOSD Attacks:
A minority of AQP4-seronegative patients with a phenotype of NMOSD have serum antibodies against MOG. (7)
Immunotherapy for NMOSD Attack Prevention:
Acute attacks are often treated with high dose intravenous methylprednisolone (1 gram daily for 3- 5 consecutive days). For patients unresponsive to glucocorticoids, then therapeutic plasma exchange is the suggested rescue treatment. (8, 9)
Premedication and Duration of NMOSD Therapy:
For attack prevention; immunotherapy is indicated as soon as the diagnosis of NMOSD is made. (10) They include treatment with eculizumab (900 mg weekly for the first four doses, followed by maintenance dosing of 1200 mg every two weeks beginning at week five), inebilizumab which is given by IV infusion as an initial dose of 300 mg, followed 2 weeks later by a second 300 mg dose. Thereafter, inebilizumab is given as 300 mg infusion every six months), rituximab (375 mg/m2 every week for 4 weeks, followed by a maintenance phase of rituximab as two 1000 mg doses separated by two weeks at 24 and 48 weeks), satralizumab (subcutaneous injection with a loading dose of 120 mg at weeks 0, 2 and 4, followed by a maintenance dose of 120 mg every four weeks), or tocilizumab (8 mg/kg every four weeks). (11, 12, 13)
Management of Refractory NMOSD Cases:
Patients on eculizumab, rituximab, inebilizumab should be pre-medicated with methylprednisolone 100 mg, an antihistamine (diphenhydramine 25-50 mg), and an antipyretic (paracetamol 1 gm) prior to every infusion to avoid infusional reactions. Therapy is usually continued for at least five years. (14, 15) Refractory cases may benefit from other immunosuppressive agents such as, azathioprine, oral glucocorticoids or mycophenolate mofetil with no proven efficacies from clinical trials. (16)
References
1- Gault F. De la neuromyélite optique aiguë, Lyon 1894Goulden C. Optic neuritis and myelitis. Ophthal Rev 1914; 34:193.
2-Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurol Res 2006; 28:348.
3- Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum
disorders. Neurology 2015; 85:177–89.
4-Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015; 72:81.
5-Jacob A, McKeon A, Nakashima I, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry 2013; 84:922.
6-Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014; 71:276.
7-Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014; 82:474.
8-Sherman E, Han MH. Acute and Chronic Management of Neuromyelitis Optica Spectrum Disorder. Curr Treat Options Neurol 2015; 17:48.
9-Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of Neuromyelitis Optica: Review and Recommendations. Mult Scler Relat Disord 2012; 1:180.
10-Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010; 17:1019.
11-Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2020; 19:298.
12-Poupart J, Giovannelli J, Deschamps R, et al. Evaluation of efficacy and tolerability of first-line therapies in NMOSD. Neurology 2020; 94:e1645.
13-Yamamura T, Kleiter I, Fujihara K, et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N Engl J Med 2019; 381:2114.
14- FDA approves new therapy for rare disease affecting optic nerve, spinal cord. https://www.fda.gov/news-events/press-announcements/fda-approves-new-therapy-rare-disease-affecting-optic-nerve-spinal-cord (Accessed on June 15, 2020).
15- Palace J, Leite I, Jacob A. A practical guide to the treatment of neuromyelitis optica. Pract Neurol 2012;12:20914.
16-Fujihara K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol 2019; 32:385.